Module 6 lab 4: NEST
This work is licensed under a Creative Commons Attribution-ShareAlike 3.0 Unported License. This means that you are able to copy, share and modify the work, as long as the result is distributed under the same license.
Authors: Veronique Voisin, Ruth Isserlin, Chaitra Sarathy, Fatema Zohora and Gregory Schwartz
Cell-Cell Communication (CCC) in spatial transcriptomics using NEST
The presentation and processing of spatial transcriptomics is out of scope for this lab. Please refer to the CBW Spatial Transcriptomics workshop or to this review article or this one for additional information.
This lab uses examples from the 10X Visium technology

NEST (NEural network on Spatial Transcriptomics)
NEST reference paper (bioRXIv): Spatially-mapped cell-cell communication patterns using a deep learning-based attention mechanism:
Cells can communicate in 3 ways through direct contact, local chemical signaling or long-distance hormonal signaling. Paracrine signaling acts on nearby cells, endocrine signaling uses the circulatory system to transport ligands, and autocrine signaling acts on the signaling cells.
Cell-cell communication (CCC) between neighbouring cells occur via soluble signals. Cells utilize a system of surface-bound protein receptors and ligand pairs to communicate. The ligand from Cell A (source) will bind to the receptor of Cell B (target). It will trigger a signaling cascade that helps Cell B to adapt to its environment.
Spatial transcriptomics offers an advantage for studying cell-cell communication as it preserves cellular neighborhoods and tissue microenvironments.
The goal of NEST is to predict probable cell cell communication interactions using a deep learning approach. It uses ligand-receptor pairs information and NEST goal is to discover re-occuring CCC patterns in the data.
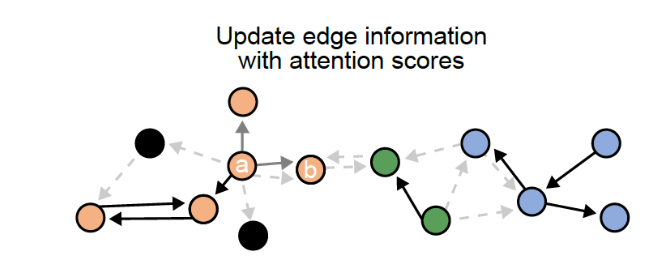
It uses a graph attention network (GAT) paired with an unsupervised contrastive learning approach to decipher patterns of communication while retaining the strength of each signal. It then uses Depth first Search (DFS) to define subgraphs to be retained after filtering the top edges using the attention score from GAT.
Input data:
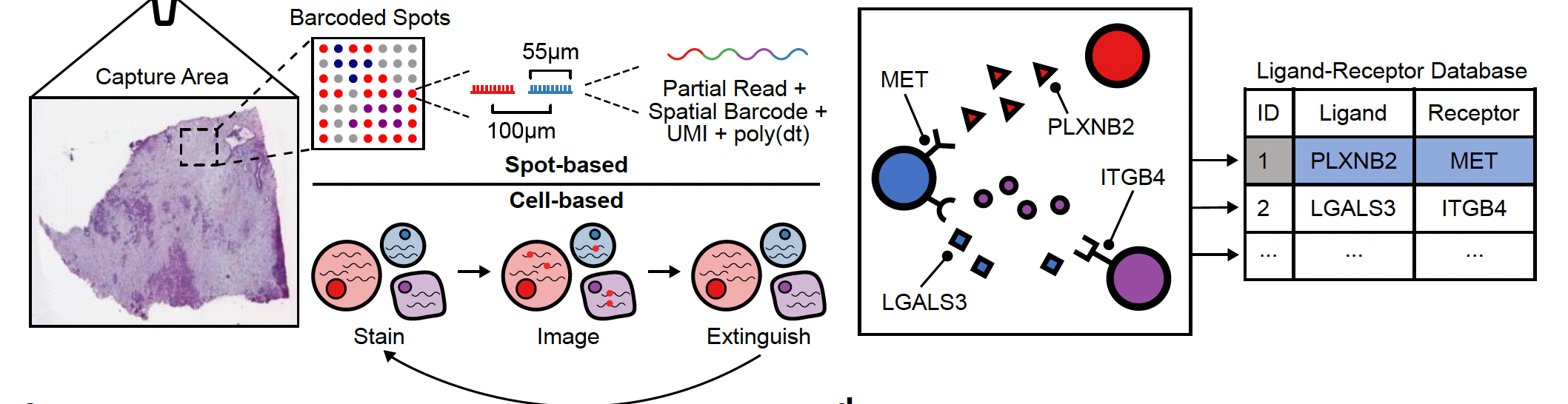 NEST needs two types of information as input data:
NEST needs two types of information as input data:- The first is the transcriptomics data with the spatial information from our biological sample (left side) and is composed of a feature matrix containing the gene expression raw counts and the postion matrix of the cells or spots.
- The second is a database of all known ligand-receptor pairs. This database is precomputed by NEST.
Step1:
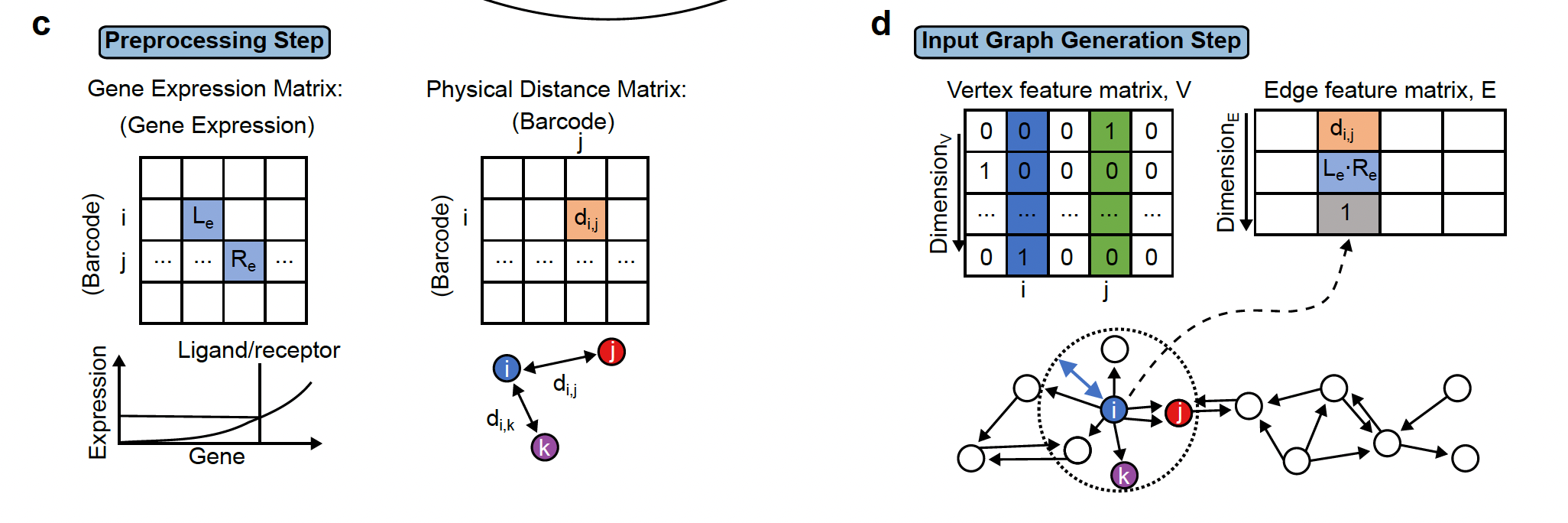 The first step is preprocessing step which includes filtering cells/spots and quantile normalization.
The first step is preprocessing step which includes filtering cells/spots and quantile normalization.Step2: Two important pieces information are collected:
- The physical distance between all cells are collected and if 2 cells are close to each other, they are linked by an edge in the resulting network.
- the presence of ligand-receptor interaction for each pair of cells. The graph (network)connects all cells that are physically close and this edge stores the ligand-receptor information between the 2 cells.
Step3:
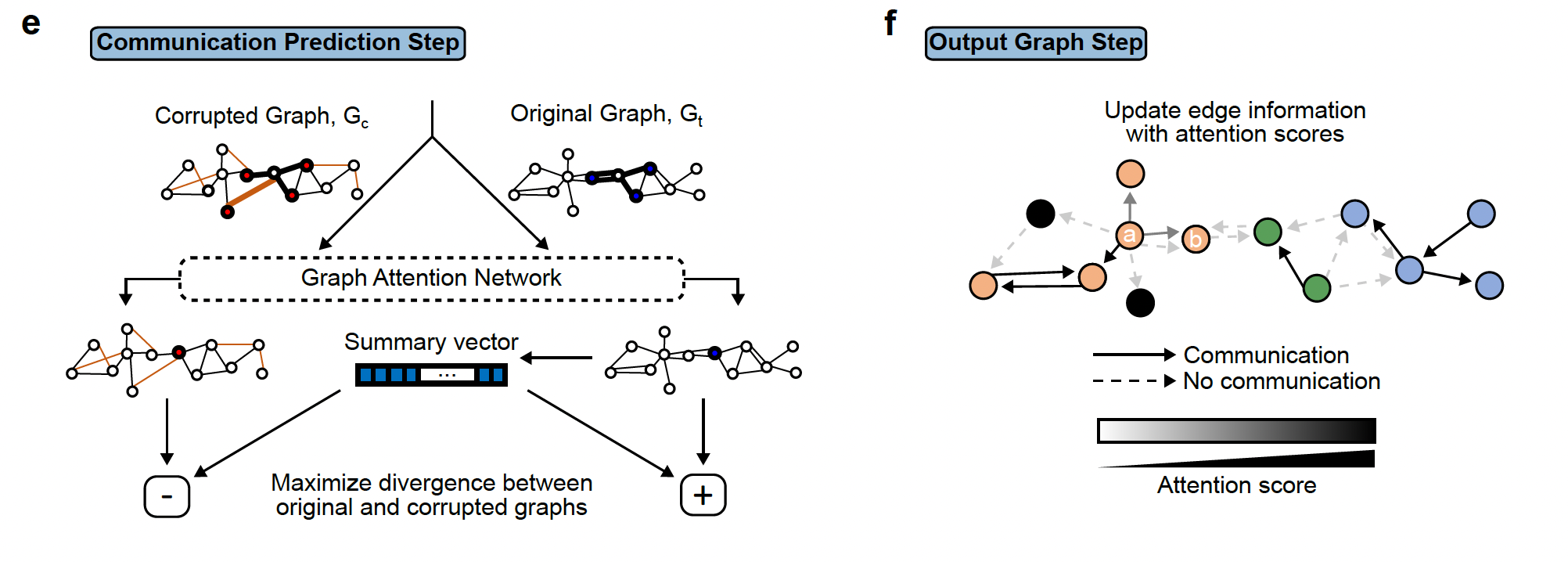 The third step involves the deep learning step that will output the final graph. The final graph retains only the edges that pass a certain threshold of the attention score. Top 20 edges are retained by default. This graph is then divided into subgraphs by the DFS algorithm. The subgraphs are represented by different colors and can be interpreted as regions of cells that are communicating a lot.
The third step involves the deep learning step that will output the final graph. The final graph retains only the edges that pass a certain threshold of the attention score. Top 20 edges are retained by default. This graph is then divided into subgraphs by the DFS algorithm. The subgraphs are represented by different colors and can be interpreted as regions of cells that are communicating a lot.Step4:
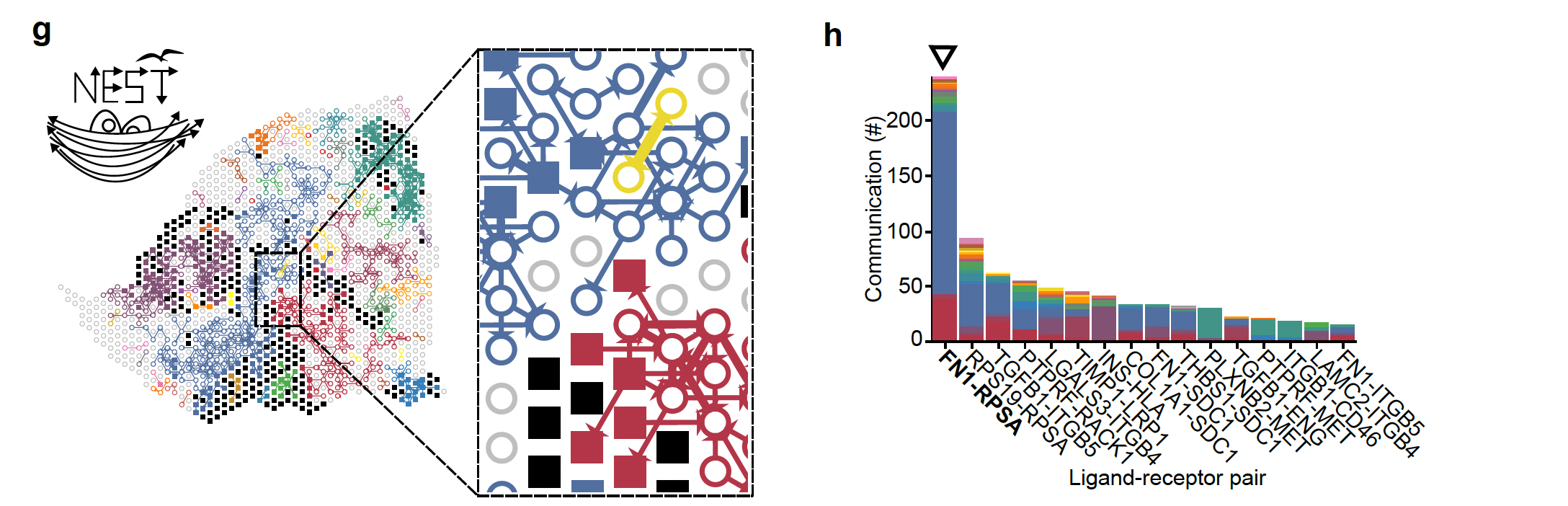
The last step is the visualization of the results of the final graph with all the ligand-receptor pairs that are the most probable cell cell communication interactions in the data under study. This is the step that we are going to explore in the lab using the NEST-interactive tool.
On the left, we see the reconstruction of the tumor section (Visium output)), the squares represent tumor cells and open circles represent stromal cells and the arrows represent the communication between the cells (ligand-receptor pairs). The different colors represent the subgraphs from the final graph of step 2. On the right, we see the histogram representing the top 20% ligand-receptor pairs that are the most represented in this dataset and evaluated by NEST and the colors are related to the subgraphs.
How to run NEST
NOTICE!
Do not run this part during the workshop.
NEST requires a graphical processing unit (GPU) to run and it is best to run it on a supercomputer (cluster). Running time and memory usage depend on the input data size.
NEST was run with 79,795 edges (each representing a ligand-receptor pair) and 1,406 vertices (each representing a Visium spot), and took 5 hours with 2.44 GB memory for each run. NEST is typically run 5 times.
Below is the information to be able to run NEST on your own after the workshop. This information is taken from the NEST github page.
NEST is written in the python language.
NEST is available as a Singularity image. Similar to Docker, it enables easy usage of NEST without set up of required environment and python packages. Furthermore, Singularity is usually installed on supercomputer/cluster system.
Steps that you would follow to run NEST:
- Step1:
- Login to your cluster system and create a folder that will store all NEST input and output data.
- Check that Singularity is installed on the cluster; check that cluster node is connected to internet
- pull the NEST singularity image
- all instructions are listed here: https://github.com/schwartzlab-methods/NEST/blob/main/vignette/running_NEST_singularity_container.md
mkdir nest_container
cd nest_container
singularity pull nest_image.sif library://fatema/collection/nest_image.sif:latest
#First time running NEST, go to NEST directory and run:
sudo bash setup.shStep2: prepare your input data. NEST takes 2 inputs:
ligand-receptor database: The default database provided by the model is a combination of the CellChat and NicheNET databases, totaling 12,605 ligand-receptor pairs. You can upload your own custom database if you are working with a different model organism.
a spatial transcriptomic data set containing:
the spatial data that contains the image and the spot localization
the feature matrix that contains the gene expression in each spot (in h5 format)

NEST requires the position matrix (tissue_position_list.tsv) and the feature matrix file. If you are working with Visium 10x, you can simply give the path to the space ranger output folder to run NEST. If you are working with other technologies, you can simply look at the format of the position and feature matrices and use this format as NEST input with your own data.
- Step3: running NEST
Preprocess
nest preprocess --data_name='V1_Human_Lymph_Node_spatial' --data_from='data/V1_Human_Lymph_Node_spatial/'Train the model
nohup nest run --data_name='V1_Human_Lymph_Node_spatial' --num_epoch 80000 --model_name='NEST_V1_Human_Lymph_Node_spatial' --run_id=1 > output_human_lymph_node_run1.log &
nohup nest run --data_name='V1_Human_Lymph_Node_spatial' --num_epoch 80000 --model_name='NEST_V1_Human_Lymph_Node_spatial' --run_id=2 > output_human_lymph_node_run2.log &
nohup nest run --data_name='V1_Human_Lymph_Node_spatial' --num_epoch 80000 --model_name='NEST_V1_Human_Lymph_Node_spatial' --run_id=3 > output_human_lymph_node_run3.log &
nohup nest run --data_name='V1_Human_Lymph_Node_spatial' --num_epoch 80000 --model_name='NEST_V1_Human_Lymph_Node_spatial' --run_id=4 > output_human_lymph_node_run4.log &
nohup nest run --data_name='V1_Human_Lymph_Node_spatial' --num_epoch 80000 --model_name='NEST_V1_Human_Lymph_Node_spatial' --run_id=5 > output_human_lymph_node_run5.log &Postprocess the model output
nest postprocess --data_name='V1_Human_Lymph_Node_spatial' --model_name='NEST_V1_Human_Lymph_Node_spatial' --total_runs=5 Please follow the NEST github page for complete instructions and vignette to run NEST
We are going to visualize the result using NEST-interactive but please note that a command line for visualization is also available in NEST:
nest visualize –data_name=‘V1_Human_Lymph_Node_spatial’ –model_name=‘NEST_V1_Human_Lymph_Node_spatial’
Practical lab : Pancreatic Ductal Adenocarcinoma (PDAC)
- PRESENTATION OF THE DATA:
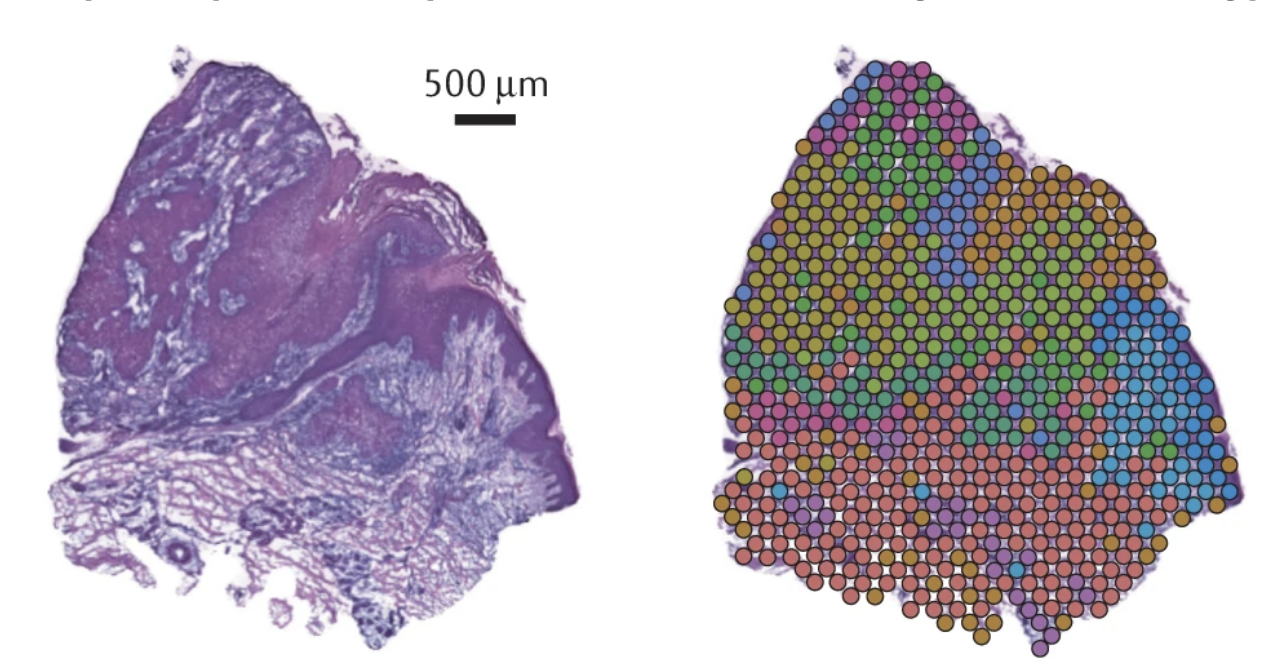
For this practical, we are working with PDAC and a tissue from a patient, PDAC_64630 , measured by Visium 10X. PDAC is recognized as a highly aggressive disease. There is immense transcriptional diversity defining discrete “Classical” and “Basal” subtypes. A PDAC tumor microenvironment is heterogeneous and consists of tumor, stromal and immune cells.
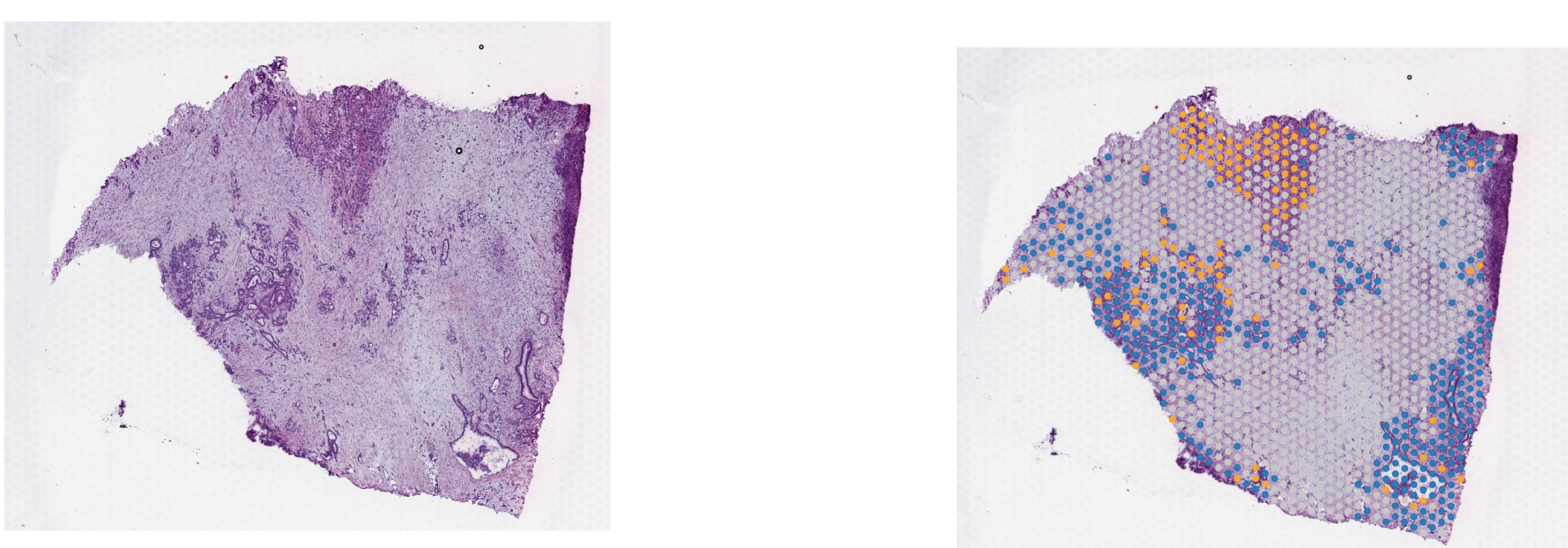
On these images, we can see the tissue section with the H&E stain on the left and we can see the Visium output on the right. The tumor regions were labelled classical (blue) and basal (red) based on some gene markers. In the middle of the tissue section, regions of stroma are colored in grey.
Goal and learning objective: - Learn how to run NEST-interactive and how to make biological inferences from the cell cell communication graph coming from the NEST output. - We will explore cell cell communication subgraphs that are localized to different regions of the tissue section: stroma, classical or basal regions. - We will explore some specific ligand-receptor pairs.
- LAUNCH THE DOCKER:
Open docker desktop (If docker is already running you can find the docker icon in your task bar. Right click on the icon and select “Go to Dashboard”).
We are going to run the Docker image that you have installed during the prework .
Open a terminal window and type the command below to launch NEST interactive:
docker run -p 8080:8080 -p 8000:8000 risserlin/nest_docker:pancreatic- Open a web browser and go to http://localhost:8080/HTML%20file/NEST-vis.html
Adjust the window size or zoom out if necessary.
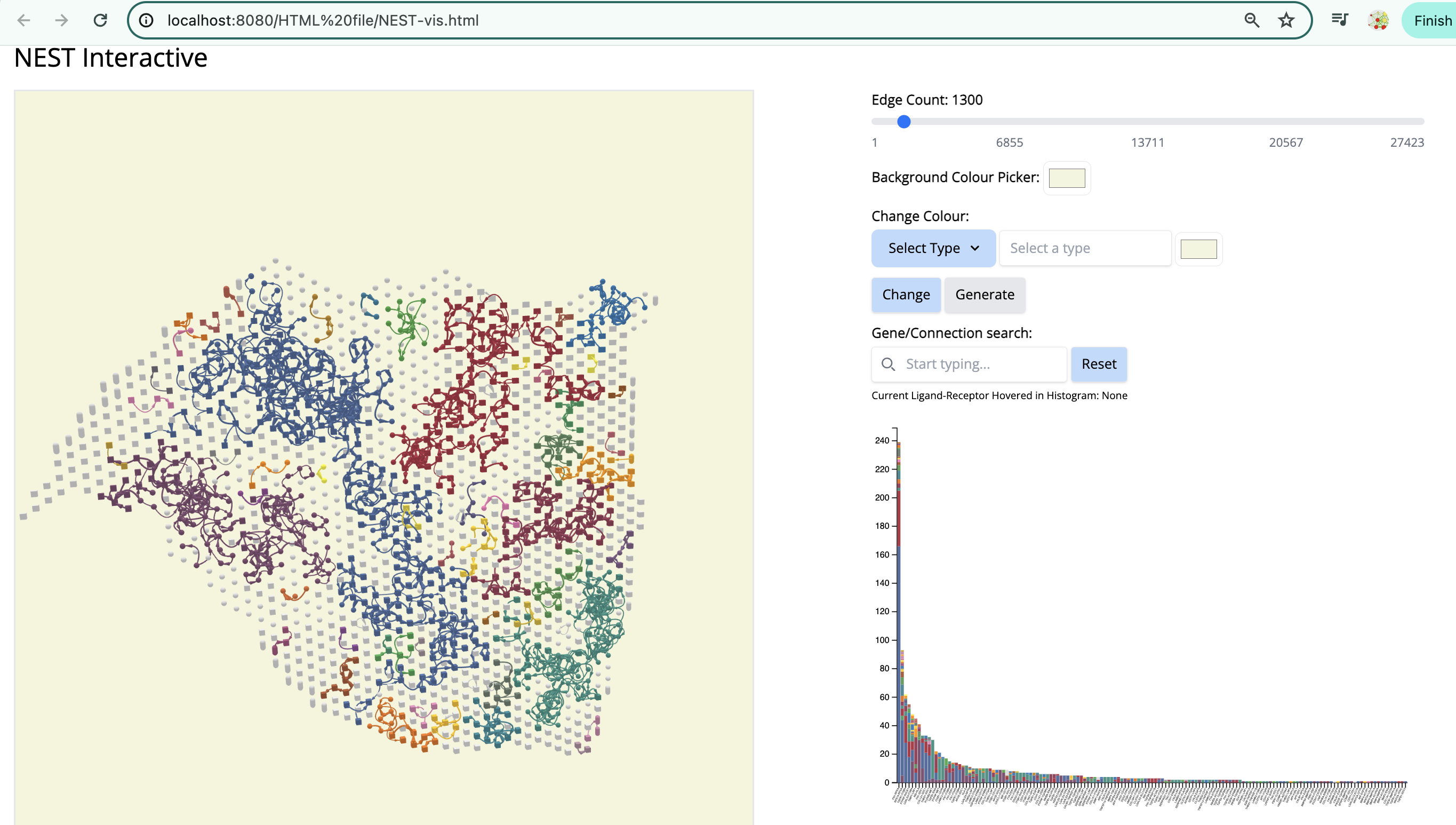
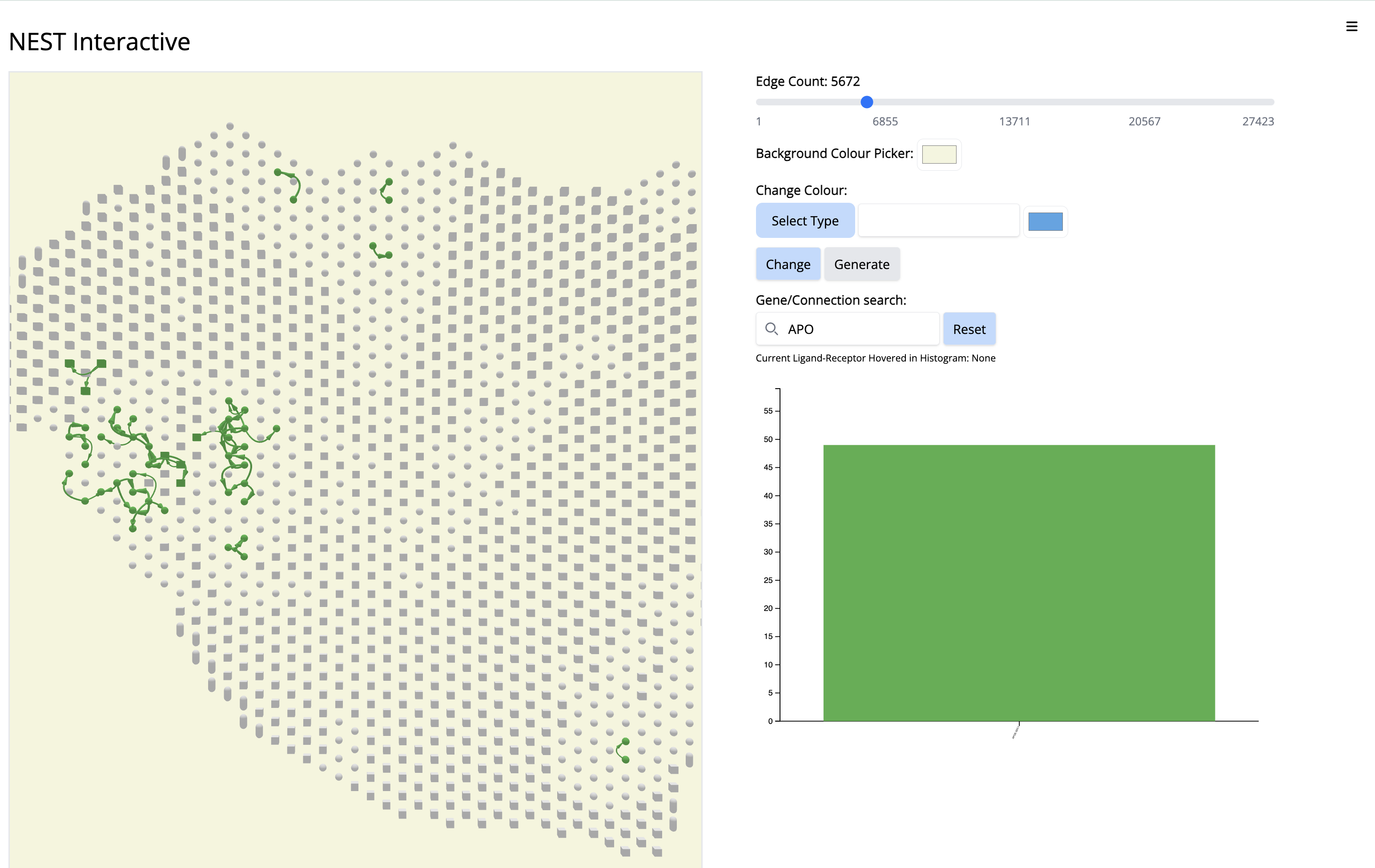
We see the Visium output of the tumor section on the left. The grey circles represent the tumor spots and the squares represent the stroma spots. Only the top 1300 edges which are the top ligand-receptor pairs based on the association score are shown. The different colors of the graph represent the different subgraphs computed by the last step of NEST ((DFS). Each subgraph groups cells that are communicating a lot together.
On the right, the histogram represents the frequency of each ligand-receptor pair on the graph. A ligand-receptor can be present in different subgraphs (represented by different colors).
- STEPS TO FOLLOW:
- Change color by vertex type: tumor - red. - Select ‘Vertex Type’ to ‘tumor’ and change the color to red. Click on ‘Change’.
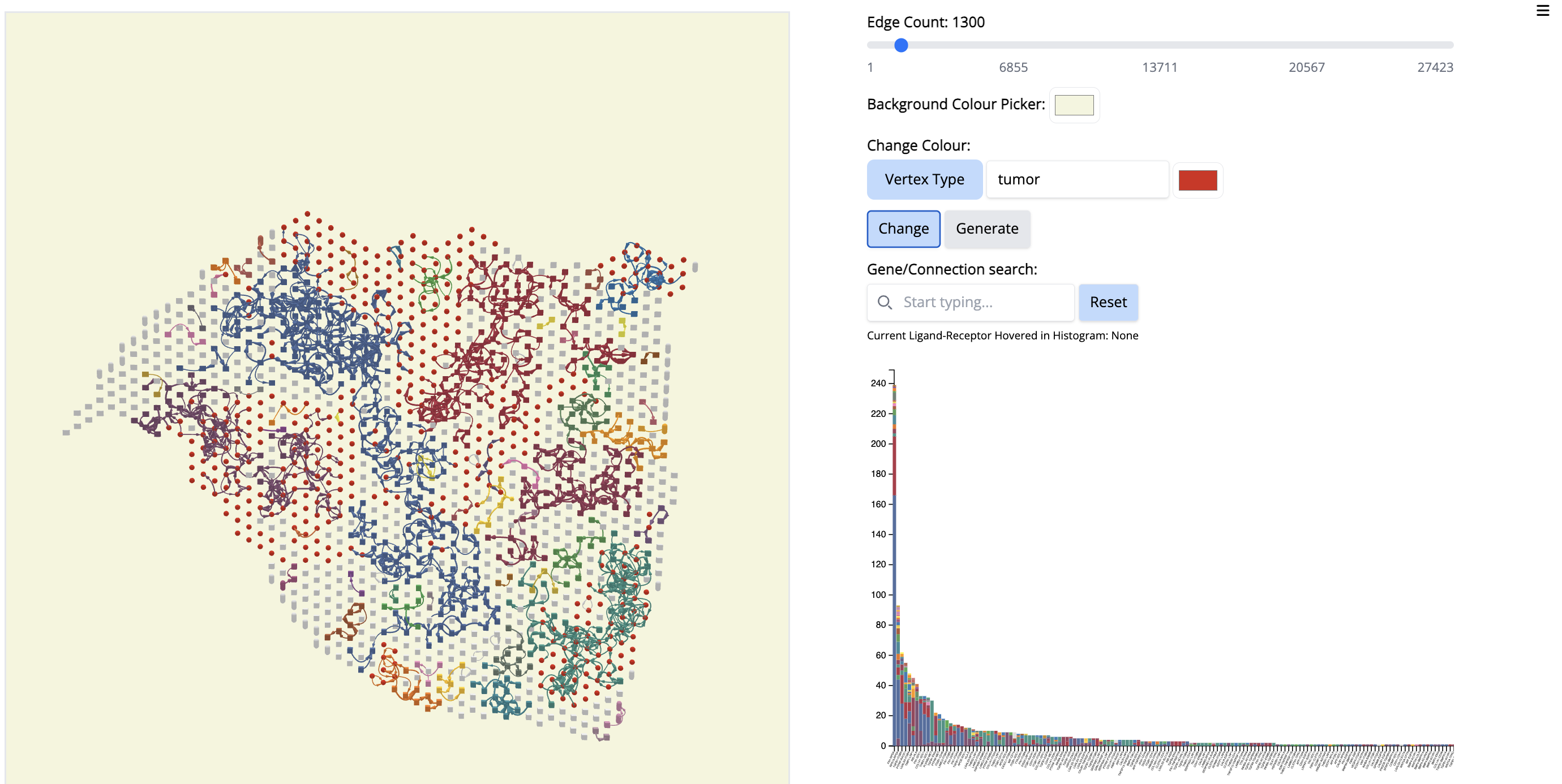
- Click on the first signal on the histogram plot. What is the first signal? Look at the literature to interpret the condition.
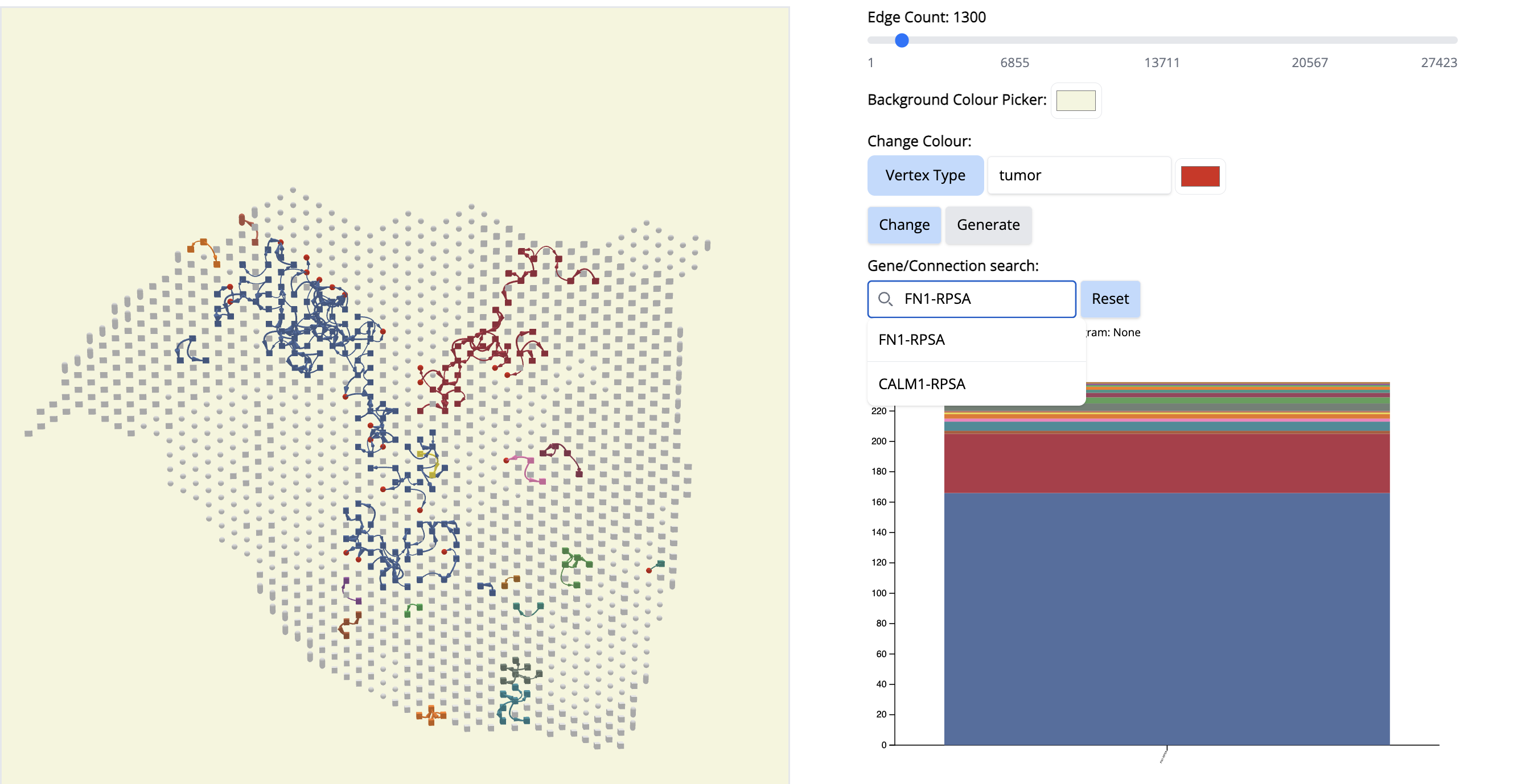
Answer: –The first signal is FN1. Fibronectin (FN1) is considered one of the main extracellular matrix constituents of pancreatic tumor stroma. High stromal FN1 expression associated with more aggressive tumors in patients with resected PDAC. Likewise, the cell membrane receptor Ribosomal Protein SA (RPSA) regulates pancreatic cancer cell migration. – so anticipate what is happening.
Reset. (Click on the ‘Reset’ button)
See which components cover a particular cancer region. Let’s pick component 10 (Cyan color).
- In the ‘Change Colour’ box, select ‘Component’, enter 10 and pick the cyan color. Click on ‘Change’.
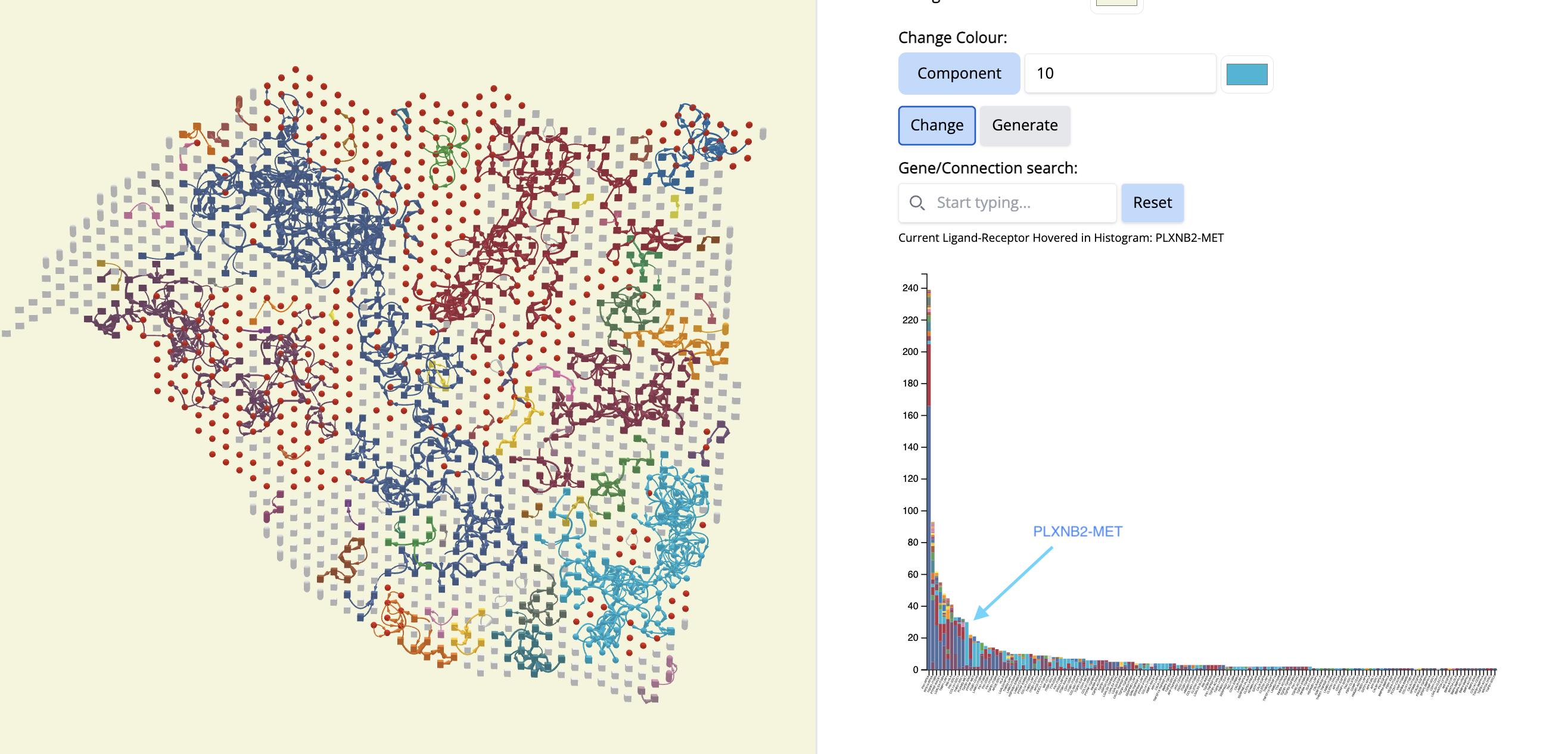
- What is remarkable is that this CCC subgraph colocalizes with the Classical subtype.
- Now, let’s see which CCCs are happening there in component 10.
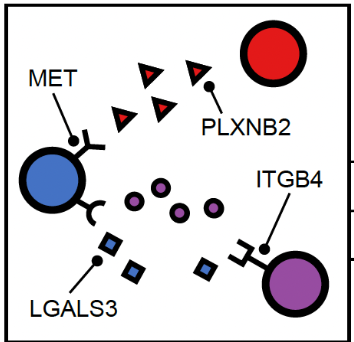
- We go to the histogram plot and click on the histogram which has the same color as component 10. Let’s pick the first most abundant CCC: PLXNB2-MET (most abundant because a bigger proportion of this CCC is associated with component 10).
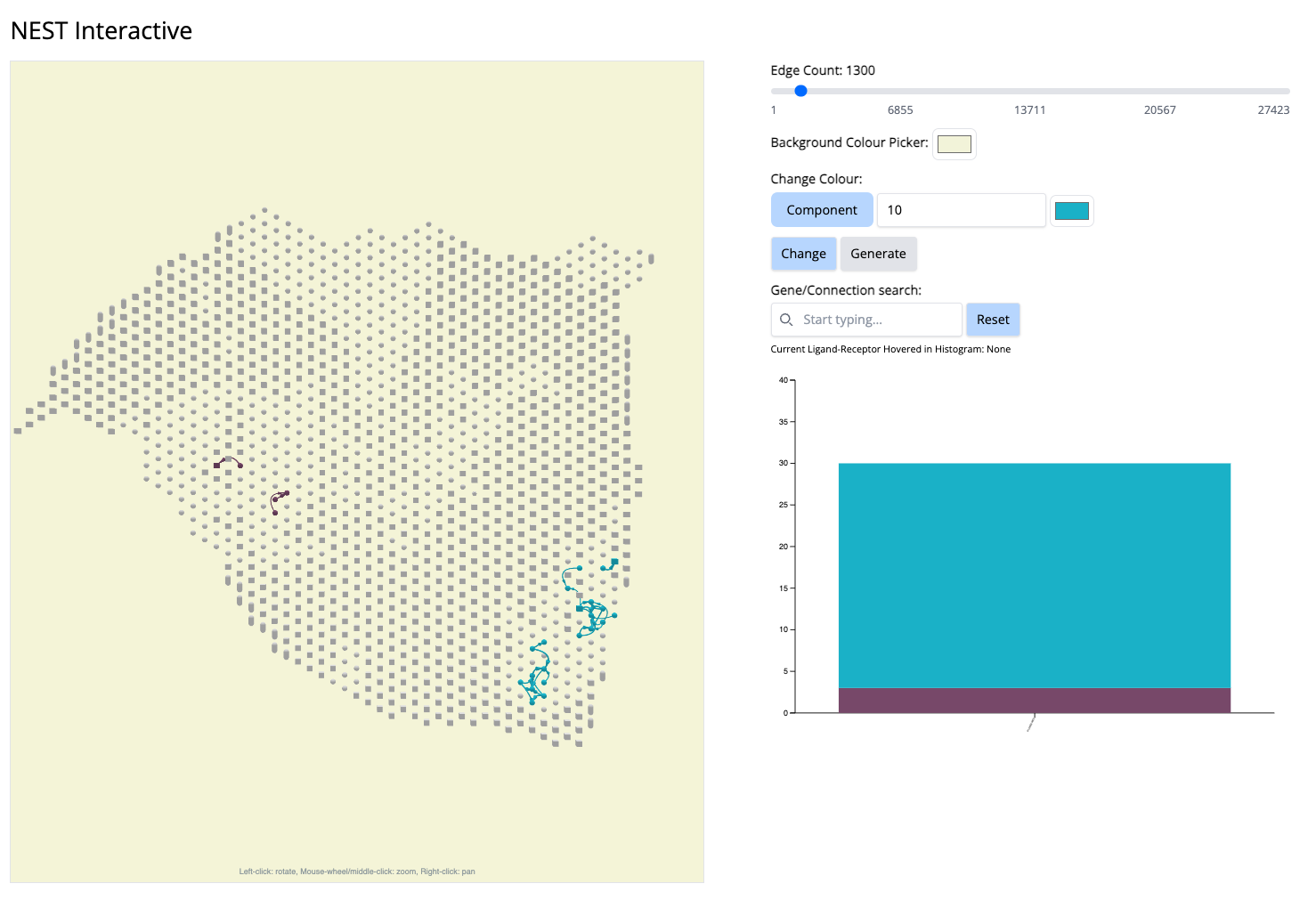
If we click on this histogram, it will show the regions where only that CCC is happening. And we see that it is happening only at that particular location. It aligns with Classical subtype of the PDAC cancer. That means, PLXNB2-MET may be a potential biomarker CCC for this subtype. → Next step for your research starting from this hypothesis: navigate further studies, e.g., comparing across multiple samples to see if PLXNB2-MET is also found in other samples in the Classical region.
Reset. (Click on the ‘Reset’ button)
Pick another cancer region - Component 4. To focus on this, let us change the color ‘by component’.
- In the “Change Colour” box, select ‘Component’, enter 4 and pick the cyan color. Click on ‘Change’.
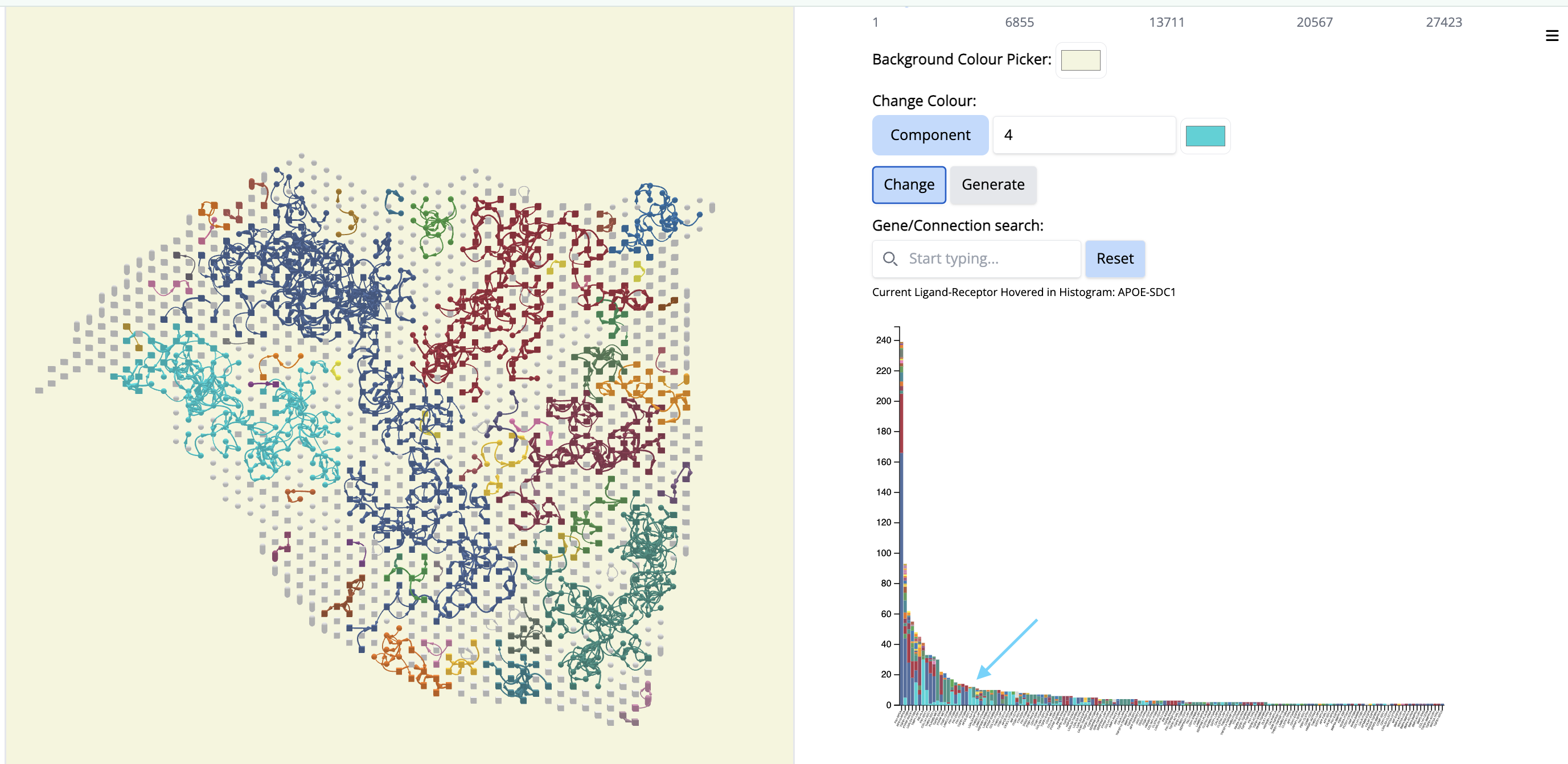
It colocalizes to another classical region of the tissue section but it will contain different ligand-receptor interactions.
- Go to the histogram plot. Pick a CCC that happens only in Component 4 - even if it is low - APOE-SDC1. Select that histogram and look at the spatial location. It is happening only in this particular region.
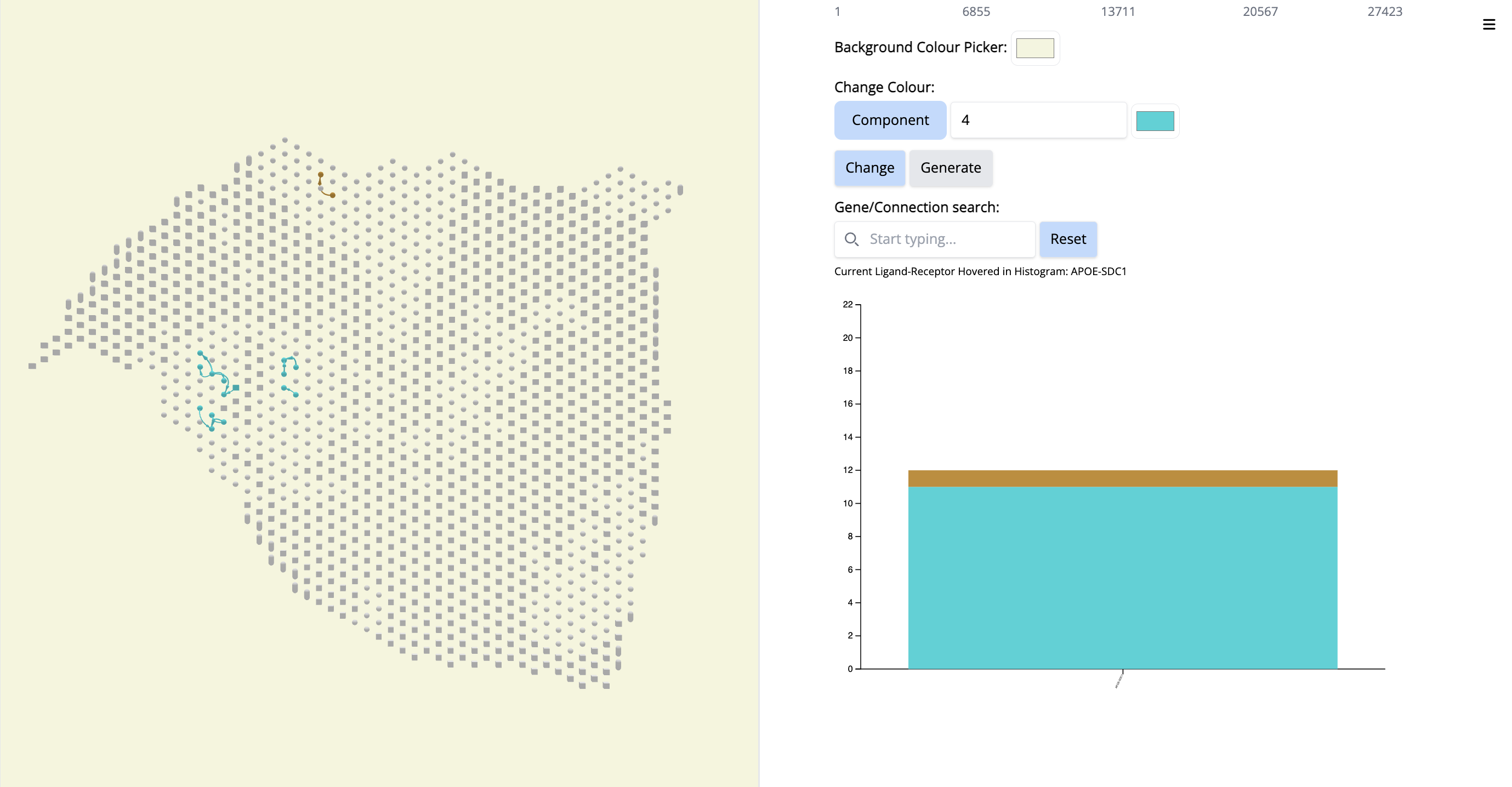
Since this interaction pair is in low amount, to gain more confidence, we could have increases the number of top CCC edges - 5000 (sliding bar on top) and repeat the process.
- Increase the number of edges. Wait until NEST_interactive finishes. In this step, NEST is recalculating the subgraphs.
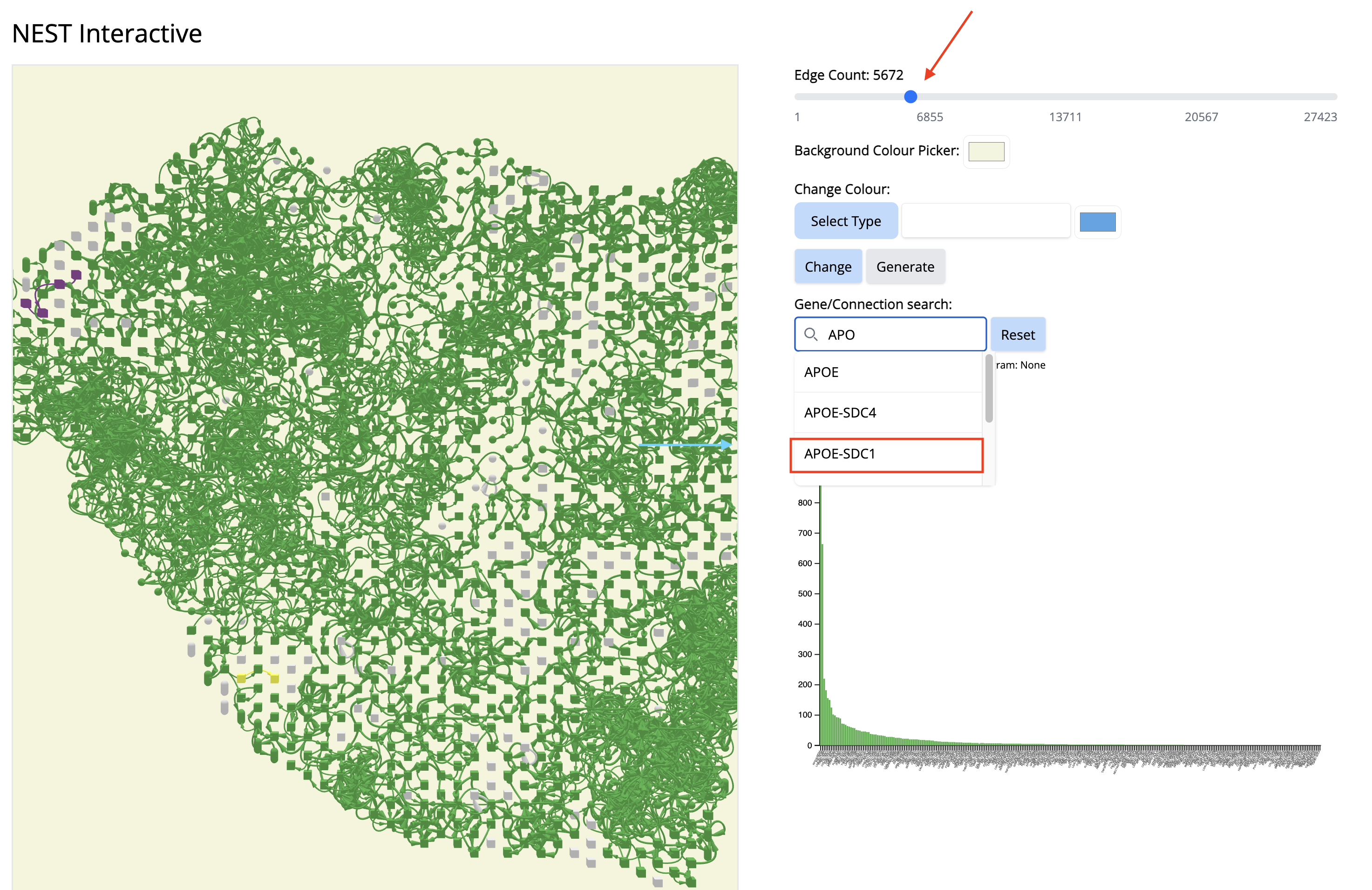
- In the ‘Gene/Connection search’ search box, look for and select ‘APOE-SDC1’